НУВІК/NUWIQ®
 від 2 °С до 8 °С. Не заморожувати
від 2 °С до 8 °С. Не заморожувати 



ІНСТРУКЦІЯ
для медичного застосування лікарського засобу
НУВІК
( NUWIQ®)
Склад:
діюча речовина:симоктоког альфа (рекомбінантний фактор коагуляції крові VIII);
1 флакон порошку для розчину для ін’єкцій містить симоктокогу альфа (рекомбінантний фактор коагуляції крові VIII) 250 МО або 500 МО, або 1000 МО, або 2000 МО;
допоміжні речовини: натрію хлорид; цукроза; L-аргініну гідрохлорид; кальцію хлорид, дигідрат; полоксамер 188; натрію цитрат, дигідрат.
Розчинник: вода для ін’єкцій.
Лікарська форма. Порошок та розчинник для розчину для ін’єкцій.
Основні фізико-хімічні властивості:
Порошок: біла грудка. Можлива невелика кількість білого порошку.
Розчинник: прозора, безбарвна рідина, без часток.
Фармакотерапевтична група
Антигеморагічні засоби. Фактор згортання крові VIII.Код АТС B02B D02.
Фармакологічні властивості
Фактор коагуляції крові VIII зв’язується із фактором Віллебранда в кровообігу пацієнта. Активований фактор VIII діє як кофактор для активованого фактора IX, зменшуючи час перетворення фактора X в активований фактор X. Активований фактор X перетворює протромбін на тромбін. Після чого тромбін перетворює фібриноген на фібрин і утворюється згусток.
Гемофілія А - спадкове захворювання крові, пов’язане із статтю, спричинене вродженим дефіцитом фактора згортання VIII:C, що призводить до крововиливів у суглоби, м’язи, внутрішні органи, які виникають спонтанно або в результаті випадкової чи хірургічної травми. При замісній терапії рівні фактора VIII в плазмі збільшуються, завдяки чому тимчасово корегується дефіцит фактора згортання крові VIII і схильність до кровотеч.
Популяція дорослих і пацієнтів віком 12 - 65 років.
Профілактика: В клінічному дослідженні у 32 дорослих пацієнтів з тяжкою гемофілією А середня профілактична доза лікарського засобу Нувік становила 468,7 МО/кг/місяць. Лікування кровотечі: Середня доза для лікування кровотечі у пацієнтів, що отримували профілактику, становила 33,0 МО/кг. В іншому клінічному дослідженні 22 дорослих пацієнти отримали лікування за потребою. Всього в 986 випадках кровотеч було застосовано середню дозу 30,9 МО/кг. Загалом, незначні кровотечі потребували менших, а сильні кровотечі потребували втричі більших середніх доз.
Індивідуалізована профілактика: Індивідуалізована профілактика, що базується на ФК, оцінювалася у 66 дорослих пацієнтів, які раніше отримували лікування (PTPs), із тяжкою гемофілією А. Після 1-3 місячного стандартного етапу профілактики (отримання дози через день або 3 рази на тиждень), 44 (67 %) пацієнти перейшли на режим дозування, що базується на оцінці їх ФК, а 40 пацієнтів завершили 6-місячну профілактику відповідно до призначених доз і схеми лікування. Із цих пацієнтів, 34 (85%) отримували лікування два рази на тиждень або менше. 33 (82,5%) пацієнтів не мали ніяких кровотеч, а 36 (90,0%) пацієнтів не мали спонтанних кровотеч. Середня+ SD аннуілізована (розрахована на річній основі) частота кровотеч складала 1,2+ 3,9, а середня+ SD доза становила 52,2+ 12,2 МО/кг на ін’єкцію та 99,7+ 25,6 МО/кг на тиждень.
Слід відмітити, що аннуалізована частота кровотеч (ABR) не є порівнянною між різними концентратами фактору та між різними клінічними дослідженнями.
Педіатрична популяція
Дані отримано у 29 раніше лікованих дітей віком від 2 до 5 років, 31 дитини віком від 6 до 12 років і одного підлітка віком 14 років. Середня профілактична ін’єкційна доза становила 37,8 МО/кг. Двадцять пацієнтів отримали середні дози понад 45 МО/кг. Середня профілактична доза лікарського засобу Нувік на місяць становила 521,9 МО/кг. Для лікування кровотеч дітям призначали більшу середню дозу лікарського засобу Нувік (43,9 МО/кг), ніж дорослим (33,0 МО/кг), а також більшу середню дозу для лікування як малих, так і великих кровотеч (78,2 МО/кг проти 41,7 МО/кг). Дітям молодшого віку в цілому були необхідні більші середні дози (6 - 12 років - 43,9 МО/КГ; 2 - 5 років - 52,6 МО/кг). Ці дані підтверджувалися довготривалим подальшим лікарським наглядом 49 таких дітей, яких лікували протягом додаткового середнього періоду приблизно 30 місяців (діапазон від 9,5 до 52 місяців); протягом цього періоду 45% дітей не мали спонтанних кровотеч.
Дані від 108 пацієнтів, які раніше не отримували лікування, із тяжкою гемофілією А (˂ 1% FVIII:C) були отримані в проспективному відкритому клінічному дослідженні. У більшості пацієнтів профілактичне лікування було розпочато після виникнення першої кровотечі, що потребувала лікування.
Доросла популяція
Таблиця 1.
Фармакокінетичні параметри лікарського засобу Нувік (доза 50 МО/кг) у дорослих віком від 18 до 65 років з тяжкою гемофілією А, які лікувалися раніше (n).
Фармакокінетичні параметри | Хромогенний аналіз | |
Середнє ± SD | Медіана (межі) | |
AUC (год*МО/мл) | 22,6 ± 8,0 | 22,3 (8,4 - 38,1) |
T 1/2 (год) | 14,7 ± 10,4 | 12,5 (5,4 - 55,6) |
IVR (%/МО/кг) | 2,5 ± 0,4 | 2,5 (1,7 - 3,2) |
CL (мл/год/кг) | 3,0 ± 1,2 | 2,7 (1,5 - 6,4) |
AUC - площа під кривою (FVIII:C), T ½ - період напіввиведення
IVR - відновлення показниківin vivo, CL - кліренс, SD - стандартне відхилення
Таблиця 2.
Фармакокінетичні параметри лікарського засобу Нувік (доза 50 МО/кг) у дітей віком від 6 до 12 років з тяжкою гемофілією А, які лікувалися раніше (n=12).
Фармакокінетичні параметри | Хромогенний аналіз | |
Середнє ± SD | Медіана (межі) | |
AUC (год*МО/мл) | 13,2 ± 3,4 | 12,8 (7,8 - 19,1) |
T 1/2 (год) | 10,0 ± 1,9 | 9,9 (7,6 - 14,1) |
IVR (%/МО/кг) | 1,9 ± 0,4 | 1,9 (1,2 - 2,6) |
CL (мл/год/кг) | 4,3 ± 1,2 | 4,2 (2,8 - 6,9) |
AUC - площа під кривою (FVIII:C), T ½ - період напіввиведення
IVR - відновлення показниківin vivo, CL - кліренс, SD - стандартне відхилення
Таблиця 3.
Фармакокінетичні параметри лікарського засобу Нувік (доза 50 МО/кг) у дітей віком від 2 до 5 років зтяжкою гемофілією А, які лікувалися раніше (n =13).
Фармакокінетичні параметри | Хромогенний аналіз | |
Середнє ± SD | Медіана (межі) | |
AUC (год*МО/мл) | 11,7 ± 5,3 | 10,5 (4,9 - 23,8) |
T 1/2 (год) | 9,5 ± 3,3 | 8,2 (4,3 - 17,3) |
IVR (%/МО/кг) | 1,9 ± 0,3 | 1,8 (1,5 - 2,4) |
CL (мл/год/кг) | 5,4 ± 2,4 | 5,1 (2,3 - 10,9) |
AUC - площа під кривою (FVIII:C), T ½ - період напіввиведення
IVR - відновлення показниківin vivo, CL - кліренс, SD - стандартне відхилення
Педіатрична популяція
Згідно з опублікованими даними, показники відновлення та період напіввиведення були нижчими у дітей молодшого віку, ніж у дорослих, а показник виведення - вищим, що може бути частково пояснено більшим об’ємом плазми на кілограм маси тіла у пацієнтів молодшого віку.
Підгрупи за масою тіла
Таблиця 4.
Фармакокінетичні параметри лікарського засобу Нувік (доза 50 МО/кг) у дорослих віком від 18 до 65 років з тяжкою гемофілією А, які раніше лікувалися (n ), за масою тіла пацієнта
Фармакокінетичні параметри | Усі пацієнти (n ) | З нормальною масою тіла вага (n =14) | Стан перед ожирінням (n =4) | Ожиріння (n =2) |
Хромогенний аналіз, середнє ± SD | ||||
AUC (год*МО/мл) | 22,6 ± 8,0 | 20,4 ± 6,9 | 24,9 ± 8,9 | 33,5 ± 6,5 |
T 1/2 (год) | 14,7 ± 10,4 | 14,7 ± 12,1 | 13,4 ± 5,9 | 17,2 ± 4,8 |
IVR (%/МО/кг) | 2,5 ± 0,4 | 2,4 ± 0,4 | 2,7 ± 0,4 | 2,8 ± 0,3 |
CL (мл/год/кг) | 3,0 ± 1,2 | 3,2 ± 1,3 | 2,6 ± 1,0 | 1,8 ± 0,4 |
Хромогенний аналіз, медіана (межі) | ||||
AUC (год*МО/мл) | 22,3 (8,4 - 38,1) | 21,2 (8,4- 32,6) | 23,3 (17,4 - 35,5) | 33,5 (28,9 - 38,1) |
T 1/2 (год) | 12,5 (5,4 - 55,6) | 12,3 (5,4- 55,6) | 11,2 (9,3 - 22,0) | 17,2 (13,8 - 20,6) |
IVR (%/МО/кг) | 2,5 (1,7 - 3,2) | 2,4 (1,7 - 3,1) | 2,8 (2,3 - 3,2) | 2,8 (2,6 - 3,0) |
CL (мл/год/кг) | 2,7 (1,5 - 6,4) | 2,8 (1,7 - 6,4) | 2,5 (1,6 - 3,7) | 1,8 (1,5 - 2,0) |
Нормальна маса тіла BMI - 18.5 - 25 кг/м2, Підвищена маса тіла: BMI 25 - 30 кг/м2, Ожиріння: BMI > 30 кг/м2, SD - стандартне відхилення
Клінічні характеристики
Показання.
Лікування та профілактика кровотечі у пацієнтів із гемофілією А (вроджений дефіцит фактора VIII).
Препарат Нувік можна застосовувати пацієнтам усіх вікових груп.
Протипоказання.
Підвищена чутливість до активної речовини або будь-якої з допоміжних речовин лікарського засобу.
Взаємодія з іншими лікарськими засобами та інші види взаємодій.
Не проводилось ніяких досліджень щодо взаємодій лікарського засобу Нувік з іншими лікарськими засобами.
Особливості застосування.
Відстежуваність
Для поліпшення відстежуваності біологічних лікарських засобів слід чітко реєструвати назву та номер серії введеного препарату.
Специфічна активність лікарського засобу Нувік становить приблизно 9500 МО/мг протеїну.
Симоктоког альфа (фактор коагуляції крові VIII (рДНК)) - чистий протеїн, який має 1440 амінокислот. Послідовність амінокислот подібна до 90+80 кДа форми фактора
плазми людини VIII (тобто з видаленим В-доменом). Нувік отримують за технологією рекомбінантної ДНК з генетично модифікованих клітин нирки ембріона людини (НЕК) 293F. Під час виробничого процесу та до готового лікарського засобу не додаються матеріали тваринного чи людського походження.
Підвищена чутливість
Як і при застосуванні будь-якого внутрішньовенного протеїновому препарату, є ймовірність розвитку алергічної реакції та підвищеної чутливості. Нувік містить слідові кількості білків людської клітини, які відрізняються від фактора VIII. У разі появи симптомів підвищеної чутливості потрібно негайно припинити застосування лікарського засобу і звернутися за медичною допомогою. Пацієнтів необхідно проінформувати про ранні ознаки реакцій гіперчутливості, такі як кропив’янка, генералізована кропив’янка, відчуття стиснення в грудній клітці, ускладнене дихання, хрип, артеріальна гіпотензія і анафілаксія.
У разі виникнення шоку слід здійснювати стандартне медичне лікування шокового стану.
Інгібітори
Формування нейтралізуючих антитіл (інгібіторів) фактора VIII є відомим ускладненням, що виникає при лікуванні осіб із гемофілієюА. Ці інгібітори зазвичай являють собою імуноглобуліни IgG, дія яких спрямована проти прокоагуляційної активності фактора VIII, і які кількісно визначаються в одиницях Бетезда (БО) на 1 мл плазми, з використанням модифікаційного аналізу. Ризик утворення інгібіторів корелює з тяжкістю захворювання, а також залежить від впливу фактора VIII, причому він найвищий протягом перших 50 днів впливу, але триває протягом усього життя, хоча ризик є рідкісним.
Випадки повторного утворення інгібіторів (низький титр) спостерігалися при переході з одного препарату рекомбінантного фактораVIII на інший у пацієнтів, які раніше отримували лікування препаратом протягом більше 100днів та у яких в анамнезі наявне утворення інгібіторів. У зв’язку з цим рекомендується ретельний моніторинг усіх пацієнтів щодо утворення інгібіторів після будь-якої зміни лікарського засобу.
Клінічна значимість утворення інгібіторів залежатиме від титру інгібітора: інгібітори з низьким титром, які тимчасово присутні або постійно зберігають низький титр, являють собою менший ризик недостатньої клінічної відповіді, ніж інгібітори з високим титром.
Необхідний ретельний моніторинг усіх пацієнтів, які отримують лікування рекомбінантними факторами згортання кровіVIII, щодо утворення інгібіторів шляхом належного клінічного спостереження та лабораторних аналізів. Якщо очікувані рівні активності фактораVIII у плазмі крові не досягаються або якщо кровотеча не припиняється при застосуванні відповідної дози препарату, слід провести аналізи для визначення наявності інгібіторів фактораVIII. Для пацієнтів з високими титрами інгібіторів терапія факторомVIII може бути неефективною, тому потрібно розглянути інші терапевтичні варіанти, такі як індукція імунної толерантності (ITI). Лікування таких пацієнтів повинні проводити лікарі з досвідом лікування гемофілії та випадків утворення інгібіторів фактораVIII.
Серцево-судинні ускладнення
У пацієнтів із існуючими факторами ризику серцево-судинних захворювань замісна терапія FVIII може збільшити ризик серцево-судинних захворювань.
Ускладнення, пов’язані із застосуванням катетера
У разі необхідності використання пристрою для центрального венозного доступу (CVAD) слід розглянути ризик розвитку ускладнень, пов’язаних з його використанням, у тому числі місцевих інфекцій, бактеріємії та тромбозу на ділянці з катетером.
Настійно рекомендується, щоб при кожному введенні лікарського засобу Нувік записувати назву та номер партії (серії) лікарського засобу для визначення зв’язку між станом пацієнта і партією введеного лікарського засобу.
Усі застереження стосуються як дорослих, так і дітей.
Інформація щодо допоміжних речовин (вміст натрію)
1 мл розведеного розчину містить 7,35 мг (18,4 мг натрію на флакон), тобто, по суті, «не містить натрію».
Проте, залежно від маси тіла та дозування, пацієнт може отримувати більше ніж один флакон. Дану інформацію слід враховувати пацієнтам, які дотримуються дієти з контрольованим вмістом натрію.
Застосування у період вагітності або годування груддю.
Дослідження впливу Нувік на репродуктивну функцію тварин не проводилися. Оскільки гемофіліяА рідко виникає у жінок, досвід застосування Нувік в період вагітності та годування груддю відсутній. Тому Нувік можна призначати під час вагітності та годування груддю за показаннями у разі нагальної потреби. Даних щодо впливу на фертильність немає.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Не виявлено жодного впливу на здатність керувати автотранспортом та працювати з іншими механізмами.
Спосіб застосування та дози.
Терапію слід проводити під наглядом лікаря, який має досвід у лікуванні гемофілії.
Моніторинг лікування
Під час курсу лікування, рекомендується відповідне визначення рівнів фактора VIII, щоб контролювати дозу, необхідну для введення, та частоту повторних інфузій. Окремі пацієнти можуть мати різну відповідь на застосування фактора VIII, демонструючи різні періоди напіввиведення з організму та одужання (відновлення). Доза залежно від маси тіла пацієнта може потребувати підбору (коригування) у разі недостатньої або надмірної маси тіла. У разі обширного хірургічного втручання точний моніторинг замісної терапії за допомогою аналізу на згортання крові (активність фактора VIII в плазмі крові) є обов’язковим.
При використанні одностадійного аналізу коагулюючої активності крові на основі тромбопластинового часу (aPTT)in vitro для визначення активності фактора VIII в зразках крові пацієнтів на результати активності фактора VIII в плазмі крові можуть суттєво впливати як тип aPTT реагенту, так і стандартний зразок, що використовуються в аналізі. Також можуть спостерігатися значні розбіжності між результатами аналізу, отриманого за допомогою одно-стадійного аналізу коагулюючої активності крові на основі aPTT, і хромогенного аналізу відповідно до Європейської фармакопеї. Це дуже важливо, особливо при зміні лабораторії та/або реагентів, що використовуються в аналізі.
Спосіб застосування
Дозування і тривалість замісної терапії залежать від тяжкості дефіциту фактора VIII, місцезнаходження і ступеня кровотечі, а також від клінічного стану пацієнта.
Кількість одиниць фактора VIII, що вводиться, визначається у міжнародних одиницях (МО), що пов’язано з діючим стандартом концентрату ВООЗ для лікарських засобів фактора VIII. Активність фактора VIII у плазмі визначається або у відсотках (відносно нормальної плазми людини), або переважно у міжнародних одиницях (відповідно до Міжнародного стандарту для фактора VIII у плазмі).
1 міжнародна одиниця (МО) активності фактора VIII є еквівалентною кількості фактора VIII у 1 мл нормальної плазми людини.
Лікування у разі потреби
Розрахунки необхідної дози фактора VIII базуються на емпіричних даних про те, що 1 міжнародна одиниця (МО) фактора VIII на 1 кг маси тіла збільшує активність фактора VIII у плазмі крові приблизно на 2 % від нормальної активності або на 2 МО/дл. Необхідна доза визначається за допомогою такої формули:
І.
Необхідні одиниці = маса тіла (кг) × бажаний ріст фактора VIII (%) (МО/дл) × 0.5 (МО/кг на МО/дл)
ІІ.
Очікуваний ріст фактора VIII (% від нормального) = | 2 × ведені МО |
маси тіла (кг) |
II.
Доза, яку потрібно вводити, і частота введення завжди повинні бути орієнтовані на клінічну ефективність у кожному окремому випадку.
У разі нижчезазначених геморагічних ситуацій активність фактора VIII не повинна опускатися нижче даного рівня активності плазми (у % від нормального або МО/дл) у відповідний період. Таблицю 5 можна використовувати для визначення дозування у періоди кровотеч і операцій.
Таблиця 5
Ступінь кровотечі/ тип хірургічної процедури | Необхідний рівень фактора VIII (%) (МО/дл) | Частота дозування (години)/ тривалість терапії (дні) |
Кровотеча Ранній гемартроз, м’язова чи оральна кровотеча | 20-40 | Повторюйте кожні 12 - 24 години щонайменше 1 день поки кровотеча, що супроводжується болем, не буде зупинена або до повного виліковування |
Більш екстенсивний гемартроз, м’язова кровотеча або гематома | 30-60 | Повторюйте кожні 12 - 24 години протягом 3 - 4 днів або більше поки біль та гострі порушення не пройдуть |
Життєво небезпечна кровотеча | 60-100 | Повторюйте ін’єкції кожні 8 - 24 години поки загроза не зникне |
Операція Невелика операція, включаючи видалення зубів Велика операція | 30-60 | Кожні 24 години щонайменше 1 день до повного виліковування |
80-100 (до та після хірургічного втручання) | Повторюйте ін’єкції кожні 8 - 24 години до суттєвого затягнення рани, опісля терапія на щонайменше 7 днів щоб підняти активність фактору VIII з 30% до 60% (МО/дл) |
Профілактика
Для довготривалої профілактики кровотечі у пацієнтів із тяжкою формою гемофілії А зазвичай дозування складає від 20 до 40 МО фактора VIII на 1 кг маси тіла кожні 2 - 3 дні. У деяких випадках, особливо для пацієнтів молодого віку, може виникнути потреба в збільшенні дози або частоти введення препарату.
Рекомендується протягом курсу лікування визначати рівні фактора VIII для коригування дози та частоти повторних інфузій. У разі великої хірургічної операції, зокрема, необхідний точний контроль замісної терапії за допомогою визначення активності фактора VIII у плазмі. Залежно від фактора VIII окремі пацієнти можуть демонструвати різні періоди напіввиведення і відновлення. Режим дозування можна коригувати на підставі відгуку пацієнта.
Спосіб застосування є однаковим як для дорослих, так і для дітей та підлітків, проте для дітей та підлітків, можуть бути необхідні менші інтервали між застосуванням або більші дози. Немає даних щодо застосування дітям віком до 2 років.
Метод введення
Препарат Нувік призначений для внутрішньовенного застосування.
Рекомендується вводити не більше 4 мл за хвилину.
Інструкції щодо відновлення лікарського засобу перед введенням.
Порошок потрібно розводити тільки розчинником, що додається (2,5 мл води для ін’єкцій), з використанням набору для приготування розчину для ін’єкцій. Флакон потрібно обережно збовтувати до повного розчинення порошку. Після розведення розчин потрібно набрати у шприц, що містив розчинник.
Приготовлений розчин потрібно перевіряти візуально на наявність твердих частинок і ознак зміни кольору перед введенням. Розведений розчин повинен бути прозорим та безбарвним, без жодних сторонніх частинок та мати pH від 6,5 до 7,5. Не дозволяється використання непрозорих розчинів або розчинів з наявністю осаду.
Інструкції щодо підготовки і введення препарату.
1. Доведіть розчинник (вода для ін’єкцій) у попередньо заповненому шприці і порошок у закритому флаконі до кімнатної температури. Ви можете це зробити, тримаючи їх у своїх руках, допоки вони не наберуть такої самої температури, як ваші руки. Не використовуйте жодних інших методів підігріву флакона і попередньо заповненого шприца. Ця температура повинна бути під час відкривання флакона. 2. Зніміть пластикову верхню кришку з флакона із порошком, щоб відкрити центральну частину гумової пробки. Не знімайте сіру пробку чи металеве кільце навколо верхньої частини флакона (рис.1). |
Рис 1 |
3. Протріть верхню частину флакона спиртовим тампоном. Дайте спирту висохнути. 4. Зніміть паперову кришку з адаптера флакона (рис.2). Не виймайте адаптер з упаковки. |
Рис. 2 |
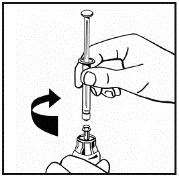
5. Поставте флакон з порошком на рівну поверхню і утримуйте його. Візьміть упаковку з адаптером і помістіть адаптер по центру гумової пробки флакона з порошком. Надавіть вниз на блок адаптера доти , доки шип адаптера на проникне в гумову пробку (рис.3). Адаптер автоматично закриється, коли ви закінчите надавлювати. |
Рис. 3 |
6. Зніміть паперову кришку з попередньо заповненого шприца. Утримуйте шток поршня на кінці і не торкайтеся стержня. Прикріпіть різьбовий кінець штока поршня до штока шприца з розчинником. Повертайте шток шприца з розчинником за годинниковою стрілкою, поки не відчуєте невеликий опір (рис.4). |
Рис. 4 |
7. Відокремте захисний пластмасовий наконечник від шприца з розчинником, натискаючи на перфорацію на ковпачку (рис.5). Не доторкайтеся до внутрішньої частини ковпачка або наконечника шприца. Якщо розчин не використовується, негайно закрийте заповнений шприц пластмасовим наконечником для зберігання з захистом від несанкціонованого доступу. |
Рис.5 |
8. Зніміть упаковку адаптера і викиньте її. 9. Щільно з’єднайте шприц з розчинником та адаптер флакона, повертаючи їх за годинниковою стрілкою доки не відчуєте незначний опір (рис.6). |
Рис.6 |
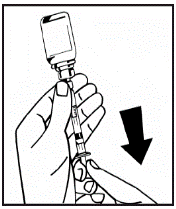
10. Повільно вливайте розчинник до флакона з порошком шляхом натискання на плунжерний стержень (рис.7). |
Рис.7 |
11. Не виймаючи шприца, обережно перемішайте або збовтайте вміст флакона декілька разів, щоб розчинити порошок. Не трясіть флакон. Зачекайте, поки весь порошок не розчиниться. 12. Візуально огляньте кінцевий розчин перед введенням. Розчин повинен бути прозорим і безбарвним, практично без видимих частинок. Не використовуйте мутні розчини або розчини, які містять видимі частинки. 13. Поверніть флакон, прикріплений до шприца, догори дном і повільно введіть кінцевий розчин у шприц (рис.8). Переконайтеся, що весь вміст флакона набрано у шприц. |
Рис.8 |
14. Від’єднайте заповнений шприц від адаптера флакона, повертаючи його проти годинникової стрілки, і викиньте пустий флакон. 15. Розчин тепер готовий до негайного використання. Не заморожуйте його. 16. Протріть місце ін’єкції спиртовим тампоном. 17. Прикріпіть набір для ін’єкцій до шприца. Введіть голку ін’єкційного набору в обрану вену. Якщо ви використовували джгут для полегшення виявлення вен, цей джгут потрібно зняти до того, як ви почнете вводити розчин. Через ризик формування фібринових згустків кров не повинна потрапляти в шприц. 18. Вводьте розчин у вену повільно, не більше ніж 4 мл за хвилину. | |
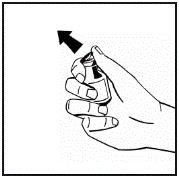

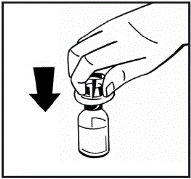



Якщо використовується більше ніж один флакон з порошком для однієї процедури, то можна використовувати ту ж саму ін’єкційну голку знову. Адаптер флакона і шприц призначені лише для одноразового використання.
Невикористаний лікарський засіб та відходи повинні бути утилізовані відповідно до місцевих вимог.
Діти.
Нувік можна застосовувати дітям будь-якого віку, але немає даних щодо застосування дітям віком до 2 років.
Передозування.
Не повідомлялось про випадки передозування.
Побічні реакції.
Резюме профілю безпеки
Гіперчутливість та алергічні реакції (які можуть включати ангіоневротичний набряк, печіння та поколювання в місці інфузії, озноб, гіперемію, генералізовану кропив’янку, головний біль, кропив’янку, артеріальну гіпотензію, сонливість, нудоту, висипання, збудженість, тахікардію, утруднене дихання, поколювання, кропив’янку, а також генералізовану кропив’янку, блювання, хрипіння/сопіння) рідко спостерігаються після препаратів фактора FVIII, але можуть в деяких випадках прогресувати до тяжкої анафілаксії (включаючи шок).
У пацієнтів з гемофілією А, які лікуються препаратом фактору VIII, а також препаратом Нувік, можуть розвиватися нейтралізуючі антитіла (інгібітори) до фактора VIII. У разі виявлення інгібіторів, причиною їх виникнення вважається недостатня клінічна відповідність у формі триваючих кровотеч або частих кровотеч, що відбуваються, незважаючи на відповідну профілактику. В таких випадках рекомендується зв’язатися з спеціалізованим центром гемофілії.
Перелік побічних реакцій у вигляді таблиці
Таблиця 6, що представлена нижче, відповідає класу системи органів MedDRA (Медичний словник нормативно-правової діяльності). Частота базувалася на звітах клінічних випробувань за участю 355 особливих пацієнтів із тяжкою гемофілією А, з яких 247 були пацієнтами, які раніше отримували лікування (PTPs) і 108 були пацієнтами, які раніше не отримували лікування ( PUPs).
Частота оцінювалася відповідно до таких критеріев: дуже часто (≥1/10); часто (≥1/100 до <1/10); нечасто (≥1/1000 до <1/100); рідко (≥1/10000 до <1/1000); дуже рідко (<1/10000), невідомо (не може бути оцінено за наявними даними).
В кожній групі частот побічні реакції представлено в порядку зменшення їх ступеня тяжкості.
Таблиця 6.
Стандартний клас систем органів MedDRA | Побічні реакції | Частота |
Порушення з боку крові та лімфатичної системи | Анемія Інгібіція фактора VIII Геморагічна анемія | нечасто* нечасто (PTPs)# дуже часто (PUPs)# нечасто* |
Порушення з боку імунної системи | Підвищена чутливість | часто* |
Порушення з боку нервової системи | Запаморочення Головний біль Парестезія (порушення чутливості) | нечасто* нечасто* нечасто* |
Порушення з боку органу слуху та рівноваги | Запаморочення | нечасто* |
Стандартний клас систем органів MedDRA | Побічні реакції | Частота |
Порушення з боку дихальної системи, органів грудної клітки та середостіння | Диспное (утруднене дихання) | нечасто* |
Порушення з боку шлунково-кишкового тракту | Сухість у роті | нечасто* |
Порушення з боку скелетно-м’язової та сполучної тканини | Біль у спині | нечасто* |
Загальні порушення та порушення в місці введення | Пірексія (лихоманка) Біль у грудях Запалення в місці ін’єкції Біль у місці ін’єкції Нездужання | часто* нечасто* нечасто* нечасто* нечасто* |
Дослідження | Позитивні антитіла, що не мають нейтралізуючої активності (у PTPs) | нечасто* |
*Підраховано як кількість пацієнтів із побічними реакціями на загальну кількість 355 досліджуваних пацієнтів, із яких 247 пацієнтів раніше отримували лікування (PTPs) і 108 пацієнтів раніше не отримували лікування (PUPs).
# Частота базується на дослідженнях із всіма препаратами фактору VIII, які включали пацієнтів із тяжкою гемофілією А.
PTPs = пацієнти, які раніше отримували лікування, PUPs = пацієнти, які не отримували раніше лікування.
Опис окремих побічних реакцій
Антитіла до антигенів фактора VIII, що не мають нейтралізуючої активності, було виявлено у одного дорослого пацієнта (див. таблицю 6). Результат був позитивним лише при розчині фактора 1, а титр антитіл був дуже низьким. Активність інгібіторів, яку вимірювали модифікованим тестом Бетезда, не була виявлена у цього пацієнта. Клінічна ефективність і in vivo відновлення лікарського засобу Нувік не вплинули на стан цього пацієнта.
Діти
Частота, тип і тяжкість побічних реакцій у дітей передбачаються такі ж самі, як і у дорослих.
Інформування про потенційні побічні реакції
Інформування про потенційні побічні реакції після реєстрації медичного препарату є важливим. Це дає змогу продовжити моніторинг балансу користі/ризику лікарського засобу. Спеціалисти галузі охорони здоров’я повинні доповідати про потенційні побічні реакції.
Термін придатності.
Порошок:2 роки.
Розчинник (вода для ін’єкцій):5 років.
Протягом усього терміну придатності, можливе зберігання лікарського засобу не більше 1 місяця при кімнатній температурі (до 25 °C). Якщо лікарський засіб вже взято з холодильника, то його неможна більше туди поміщати.
Необхідно зазначати дату початку терміну зберігання при кімнатній температурі на упаковці лікарського засобу.
Після приготування розчину для ін’єкцій хімічна і фізична стабільність була продемонстрована протягом 24 годин при зберіганні при кімнатній температурі.
З точки зору мікробіології, лікарській засіб необхідно використати одразу після приготування. Якщо його не використали одразу, то користувач лікарського засобу несе персональну відповідальність за термін придатності та умови застосування лікарського засобу.
Умови зберігання.
Порошок
Зберігати при температурі від 2 до 8 оС. Не заморожувати.
Зберігати в картонній коробці для захисту від світла.
Розчинник (вода для ін’єкцій)
Зберігати при температурі від 2 до 8 оС.
Приготовлений розчин зберігати при кімнатній температурі.
Приготовлений розчин не заморожувати.
Зберігати у недоступному для дітей місці.
Несумісність.
Через відсутність досліджень сумісності цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Потрібно використовувати лише представлені ін’єкційні набори, оскільки лікування може бути невдалим в результаті адсорбції фактора коагуляції крові VIII на внутрішніх поверхнях деяких ін’єкційних пристроїв.
Упаковка.По 250 МО, 500 МО, 1000 МО або 2000 МО порошку у флаконі; 1 флакон з порошком, 1 попередньо заповнений шприц з розчинником по 2,5 мл (вода для ін’єкцій) разом з комплектом для розчинення і внутрішньовенного введення (1 адаптер для відкриття флакона, 1 голка-метелик, 2 просочені спиртом тампони) в картонній коробці.
Категорія відпуску.
За рецептом.
Виробник.
Октафарма АБ.
Місцезнаходження виробника та адреса місця провадження його діяльності.
Ларс Форсселлс гата 23, Стокгольм, 11275, Швеція.

На сайті наведено виключно офіційні оновлені інструкції без перекладів та скорочень.
Інформація про лікарські засоби представлена на сайті для ознайомлення, не є приводом для самолікування та не є рекламою лікарських засобів.
 Важливо! До кожного лікарського засобу, який ви купуєте, обов’язково має додаватися інструкція про застосування лікарського засобу.
Важливо! До кожного лікарського засобу, який ви купуєте, обов’язково має додаватися інструкція про застосування лікарського засобу.
Тримайте всі інструкції до препаратів Домашньої аптечки під рукою – завантажуйте мобільний додаток Ліки Контроль БЕЗКОШТОВНО